Company Announcement – Majella Ryan FTOPRA

Majella Ryan FTOPRA
Ivowen are pleased to announce that Fellowship of The Organisation for Professionals in Regulatory Affairs (FTOPRA) has been conferred on Majella Ryan. According to TOPRA:
“The award of a Fellowship is intended to recognise a contribution in which the individual has used his/her professional knowledge and experience to influence, guide or develop the profession in a proactive way.
While clearly the individual’s regulatory experience and current senior position achieved are important, what is also looked for is a consistent record of activities or influences extending beyond the confines of the individual’s day-to-day activities.”
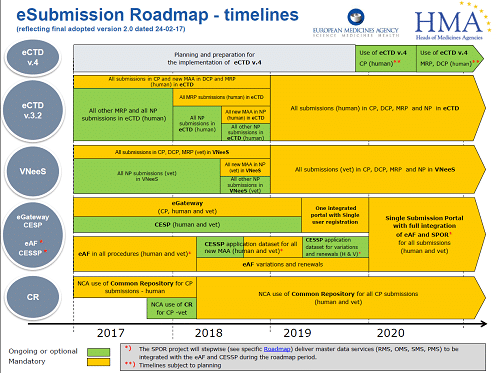
Majella Ryan has been in the regulatory affairs business for over 20 years and in this time has worked in both small and large multi-national companies, setting up Ivowen in 2002. Since then, Majella was involved in the set-up of TOPRA in Ireland, acting as the inaugural Chair in 2001/2011, as well as speaking at various conferences and providing extensive regulatory training to companies around the world. In addition, Majella is actively engaged in the publication and explanation of regulatory and guidance updates via Ivowen’s website, and via Ivowen’s EU network, EuDRAcon. As part of Ivowen’s daily work, Majella is involved in the high level regulatory strategy and support of complex projects and products like biosimilars, paediatric investigation plans and clinical trials, eCTD implementation, regulatory pathways to approval, Brexit planning, and much more.
For more information on Ivowen’s services and how we can help you, contact us.