Ivowen attended the Medicines for Europe conference in January (Regulatory and Pharmacovigilance), the annual EuDRAcon conference in May, exhibited at TOPRA in October and joined our clients from around the world at CPhI in November.
We all saw Brexit come and go, Twice !! We wait to see what lies in store for the next deadline in January 2020.
The FMD came into effect across Europe in February in most member states.
Bulgaria joined CESP, eCTD became mandatory for all human procedures, lots of new guidance was published (to keep us all on our toes) and Nitrosamines in medicinal products moved to the top of everyone’s agenda.
With the festive season now upon us and 2020 on the horizon, Ivowen are setting our sights on the year ahead.
We will be attending the Medicines for Europe conference in January 2020 (Regulatory and Pharmacovigilance) and we encourage you to contact us before mid-January with any specific questions you might like us to ‘ask the regulators’. This is a great opportunity to ask those difficult questions that you just could not get a straight answer to in 2019, on the ever present grey areas of Regulatory procedures.
To help you to plan ahead here are some helpful updates, in brief, as full articles will be posted in 2020:
Falsified Medicines Directive – Where we are now:
Implemented on 9th Feb 2019 in all MS except Greece, Italy and Belgium
The European Commission has produced a video to explain more about the safety features.
The HPRA have extended the use and learn period, initially to Sep 2019 and extended it again to end on a phased basis starting from 31st January 2020.
The MHRA is also taking a pragmatic, flexible approach to how they enforce the new legal requirements.
Nitrosamines
Step 1: MAHs should conduct a risk evaluation to identify products at risk of N-nitrosamine formation or (cross-)contamination and report the outcome by 26 March 2020 at the latest.
Ivowen are here to assist you in 2020 and will continue to provide the top quality service you have come to expect from us.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton.
https://eureg.ie/wp-content/uploads/2019/12/Hot_Air_Baloon.jpg356570ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-12-06 09:52:522023-11-06 11:08:332019 – What a year
What do you want to do next? Do you want to continue to market your product in the EU only, or are you interested in both the EU and UK markets? Read on to find out what you need to do for both scenarios.
If you would like to continue to market your product in the EU there are a few things that need to be finalised before Brexit on the 29th March 2019:
Reference Member State (RMS transfer)
This should have been done yesterday(!) The time it takes depends on the requested RMS’s workload.
Since July 2018 the MHRA is no longer accepting new applications with UK as RMS. However there are some currently authorised products wherein the UK is still the RMS.
If you have products and the UK is currently the RMS then it is vital a RMS transfer is initiated immediately. If there is only one CMS then this CMS should become the RMS (submission required). If there is more than one CMS, the preferred CMS needs to be consulted and a request sent asking them to be RMS. It is the responsibility of the MAH to secure a new RMS. The timeline for such transfers are solely dependent on the workload of the requested RMS.
Marketing Authorisation Holder (MAH) transfer
Needs to be done immediately – the time it takes is dependent on where the application is submitted.
From the 29th March 2019 the MAH for a product licensed in an EU Member State (MS) other than the UK must be based in the EU. Therefore if the MAH is currently based in the UK there needs to be a MAH transfer to one based in the EU.
If this involves purely an address change (i.e. the marketing authorisation holder remains the same legal entity but they have an address in the EU) then this is a simple type IAin (A.1).
If the new MAH is a different legal entity then the MAH transfer must follow the guidelines of the currently registered RMS and CMS, at the very least documents such as transfer agreement, proof of establishment, , power of attorney(s), Pharmacovigilance (PV) update, etc., should be in place before submission of the request.
Batch release
Needs to be done within the next 4-8 weeks unless a product is a biological / immunological product in which case submission needs to be immediate.
Products that only have batch release and quality control testing sites for finished product in the UK will have to change the batch release and testing sites for their EU products. For products that have other batch release and testing sites the MAH may choose to delete the UK site(s) or may choose to replace them. For finished products manufactured in the UK an importation site (in EEA) will need to be introduced. In many cases, a single site can perform manufacturing, testing, importation and/or batch release activities.
Needs to be done asap and in association with any MAH transfers – usually a type IA or type IAin
The Qualified Person for Pharmacovigilance (QPPV) and Pharmacovigilance Site Master File (PSMF) must be based in the EU/EEA.
If you wish to continue to market the product in the UK in addition to the EU:
The MHRA have stated that after Brexit, all currently approved authorisations will be transferred into national procedures and will remain valid.
If an application is in-progress at the time of Brexit the application will need to be submitted to the MHRA again as a national application in the case of CP procedures and that for MR or DC procedures a transitional provision will be made. HOWEVER, this is contingent on a Brexit deal that allows for a transition period. This has not yet been agreed.
To ensure the product can remain on the market / licensed, the UK are proposing the following if there is a no-deal Brexit
a MAH should be established in the UK by the end of 2020. Until then, the MHRA will require a contact in the UK. A Change of Ownership will need to be submitted to MHRA to change from an EU MAH to a UK MAH for UK MAs
the Qualified Person for Pharmacovigilance (QPPV) should be established in the UK on day one, although those without a current UK presence will have until the end of 2020 at the latest to do so, but would nevertheless be required to make arrangements for providing the MHRA with access to the relevant safety data related to UK Marketing Authorisations (MAs) at any time. Companies may choose to have the EU QPPV take on responsibility for UK MAs until the UK QPPV can be established. A variation should be submitted to the MHRA to change QPPV. Exact details of this will be consulted upon
a Qualified Person (QP) for products manufactured in the UK or directly imported into the UK from outside a country on a designated country list (whitelist) must reside and operate in the UK. A QP for products manufactured in a country on a whitelist or manufactured in a third country and imported into the UK from a country on a whitelist can reside in a country on the whitelist.
The European Union is arguably the world’s most powerful bloc and very soon it’s about to lose the United Kingdom, one of its biggest members. How and when the UK leaves the EU will have further implications that ripple around the globe.
So if you’ve heard about Brexit but haven’t been keeping up with every twist and turn of the developments, no worries! Ivowen team will provide you with everything you need to know to have your products designed for UK and Brexit affected markets authorised successfully.
What is happening?
EMA
The European Medicines Agency (EMA) will physically relocate to the Netherlands in early March 2019.
EMA will leave its premises in London on 1 March 2019.
It was confirmed that from 4 to 8 March, the Agency will operate on the basis of extended teleworking. During the course of the following week EMA staff will gradually move into the Spark building.
From 4 March 2019 onwards the official address of EMA will be that of the permanent building, located in Amsterdam Zuidas:
European Medicines Agency, Domenico Scarlattilaan 6, 1083 HS Amsterdam, The Netherlands
Meetings and visits will take place at the Spark building:
Orlyplein 24, 1043 DP Amsterdam, The Netherlands
UK guidance on Brexit
Following the outcome of the EU referendum, MHRA still feels responsible for playing a crucial role in medicines and devices regulations as well as vigilance and market surveillance.
As part of the MHRA response to exiting the EU the following Brexit guidance was issued:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
MHRA guidance on Brexit
Bearing in mind the worst-case scenario if the UK leaves the EU with no deal, the UK would no longer be part of the EU medicines and medical devices regulatory networks and consequently submissions related to human medicines would need to be submitted directly to the MHRA.
The webinar below is relevant for all pharmaceutical companies involved in making medicines regulatory submissions and vigilance activities. It also ensures that stakeholders can be informed of any IT plans and preparations. There is also a section on how all medicines related clinical trial sponsors will register and submit:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
UK legislation on medicines and medical devices
Legislation has been published which, in the event of the UK leaving the EU with no agreement, will cover the regulation of medicines, medical devices and clinical trials and allow for the continued sale. The Brexit guidance is available here:
The 2012 Regulations (as amended by the 2019 Regulations) make reference to various pieces of EU guidance, as that stood immediately before the exit day (29 March 2019).
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
EU Commission and EMA Q&As
The EU Commission & EMA have published an updated list of questions and answers related to the UKs withdrawal from the EU on the 1st February:
This confirms that dual labelling between UK & Ireland is acceptable where the labels meet the requirements of the Directive and reflect the SPC in Ireland (see Q24).
The focus of this Q&A is on the regulation of medicinal products within the centralised procedure.
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Brexit Stakeholder Event
Brexit Stakeholder Event – Ivowen was there
Following the UK’s departure from the European Union, the HPRA, together with medicines agencies in Europe, is making preparations to ensure continuity to deliver on patient and animal health remits even if the UK fully exits the current systems as scheduled. There are potential implications for the European network as a whole and particularly for Ireland with its shared marketplace, see meeting agenda below:
Contact us if you would like some more information on this event or Brexit in general
Written by Karolina Dobrychłop
https://eureg.ie/wp-content/uploads/2019/02/Brexit3.jpg437625ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-02-19 17:53:522023-11-06 11:15:36Brexit – because it affects you too…
Brexit extension date is now 31 October 2019, but there’s not time to be complacent.
Are you planning to submit a Marketing Authorisation Application (MAA) for a Non-Prescription Medicine?
If you are preparing an MAA for a Non-Prescription Medicine for a Decentralised (DCP) or Mutual Recognition Procedure (MRP) you will need to prepare a ‘Justification for Non-Prescription Classification’ document in accordance to CMDh best practice guide issued in January 2019 (CMDh/250/2012, Rev 1).
Ideally this document should be submitted as part of the initial MAA. Otherwise, you will be requested to provide it at the validation stage.
This document should be titled, “Justification for Non-Prescription Classification” and should be provided in Module 1.2. It should contain all the supporting data and evidence required to justify classification of the medicinal product as not subject to medical prescription as set out in the Guideline on Changing the Classification for the Supply of a Medicinal Product for Human Use [https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-2/c/switchguide_160106_en.pdf].
If you need any clarification or advise to prepare a justification for Non-Prescription classification report to support a Marketing Authorisation Application, Ivowen will gladly assist you in a timely manner. Please contact us.
Related news in relation to Brexit
The current Brexit extension date is 31 October 2019. The UK remains an EU Member State for the duration of the extension, with all the rights and obligations set out in the treaties and under EU law.
Nevertheless, all pharmaceutical companies in the EU are reminded to continue their preparedness for the UK’s withdrawal.
Based on the European Council decision, the deadline of 29 March 2019 referred to in Brexit related guidance should be understood as referring to 31 October 2019.
The MHRA have updated guidance on Exporting active substance manufactured in the UK in a no deal scenario on 03/06/2019.
In the event of a no deal EU exit, the UK will be recognised as a Third Country for the export of active substances for human use to the EEA.
In the event of a no deal scenario, the UK will continue to accept importation of active substances into the UK without a Written Confirmation from the same list of countries as currently (namely the European Economic Area (EEA) countries, USA, Japan, Brazil, Australia, Israel and Switzerland).
A Written Confirmation will then be required for each shipment of active substances manufactured in the UK that is exported to the EEA.
Please contact us if you need any clarification or support to supply Written Confirmation(s) for the orderly import of Active Substances, Ivowen will gladly assist you in a timely manner.
The MHRA have issued a guidance on the Handling of Active Substance Master Files and Certificates of Suitability in the event of no deal published 18 March 2019.
After Brexit, the UK will no longer participate in ASMF work-sharing procedures with EU Member States or have access to the EU Communication and Tracking System (CTS) assessment report repository. Any reference in the above guideline to the CTS ASMF assessment repository or to EU/ASMF/XXXXX reference numbers will not be applicable to UK national applications after the UK leaves the EU.
Certificates of Suitability (CEPs) are not affected by the UK leaving the EU as they are issued by the European Directorate for the Quality of Medicines and Healthcare (EDQM), which is a Directorate of the Council of Europe and a body that is independent of the EU. On leaving the EU, the UK will remain a member of the Council of Europe and a signatory to the Convention on the Elaboration of a European Pharmacopoeia.
Please contact us if you need any additional information or if you need any clarification or advise on ASMFs or CEPs, Ivowen will gladly assist you in a timely manner.
It seems that Brexit is on course for 1st January 2021….
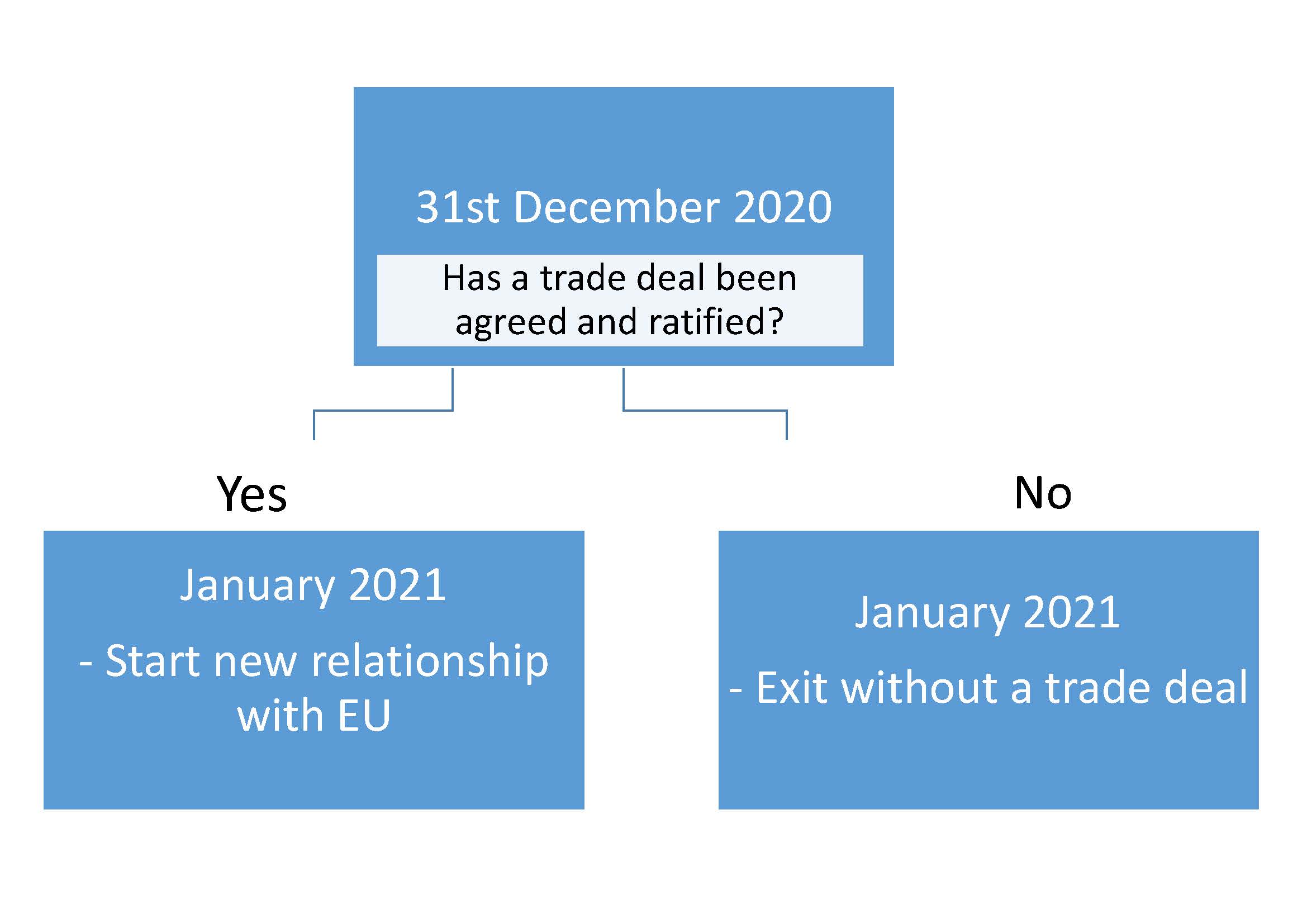
The 30th June 2020 came and the legal deadline for agreeing to an extension of the transition period has passed with no request from the UK. Therefore the Brexit trade deal transition phase will come to an end on the 31st December 2020. At this stage there will be one of two possible outcomes:The next important dates are the 15-16th October (European Council Meeting) and 26th November (penultimate plenary session of 2020. European lawmakers have stated that a trade deal must be negotiated, checked, translated and presented to the European Parliament by this date if the transition period is to end by 31 December 2020). If the UK exits without a trade deal, trade between the UK and EU will change immediately on the 1st January 2021 (i.e. Hard Brexit).
The UK Government launched a major new public information campaign to give everyone the facts that they will need to be ready for 1 January 2021. A straightforward checker tool at gov.uk/transition will help identify some of the specific steps any business or individual needs to take to be ready, and will allow companies to sign up for bespoke updates.
If you require any assistance for UK products please contact us: info@eureg.ie
Written by Emily Fletcher
Emily Flecther
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-07-22 12:42:222023-11-06 11:08:37BREXIT – No extension to the transition period
The United Kingdom has formally left the EU as of 31 January 2020 and has become what is referred to as a third country.
On 1 February 2020 a transition period started which is due to end on 31 December 2020.
During the transition period, EU pharmaceutical law will continue to be applicable to the UK, meaning that pharmaceutical companies can continue to carry out activities in the UK until the end of 2020.
Companies have until 31 December 2020 to make the necessary changes to ensure that their authorised medicines comply with EU law and can remain on the EU market.
The UK will remain on CESP for the duration of transition period (after that, if no further extension to the transition period is proposed, it will be necessary to use the MHRA portal for submissions https://pclportal.mhra.gov.uk/)
Marketing authorisation holders and applicants can still be established in the UK in 2020
Qualified Persons for Pharmacovigilance (QPPVs) and pharmacovigilance system master files (PSMFs) can still be based in the UK until the end of 2020.
Manufacturing sites, Quality control testing and Batch release sites can also still be based in the UK until the end of 2020
orphan designation holders can still be located in the UK until 31 December 2020
minor use/minor species (MUMS)/limited markets classification holders can still be located in the UK until 31 December 2020
The withdrawal agreement foresees that following its departure from the EU on 31 January 2020, the UK will no longer participate in EU institutions and their decision-making. For the CMDh this means that as of 1 February 2020, no one who represents the UK, or is appointed or nominated by the UK can systemically participate in the CMDh meetings.
During the transition period, the UK will not be able to act as RMS in MRP/DCP, but the UK can participate in MRP/DCP as CMS.
Ivowen are here to assist you with all your Brexit related needs and dossier amendments.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-02-10 11:40:492023-11-06 11:08:32BREXIT – EVERYTHING stays the same for 2020
The UK has left the EU and the transition period after Brexit comes to an end this year.
The MHRA have issued new guidance for industry and organisations effective from 01st January 2021. From this date the MHRA will be the UK’s standalone medicines and medical devices regulator.
Areas covered in the new guidance include:
Clinical Trials
From 1 January 2021, for registering clinical trials, existing and established international registers will still be used, such as ISRCTN registry (UK), or ClinicalTrials.gov (USA), to ensure the public is aware of your trial. For trials involving both UK and EU sites a record in the EU Clinical Trials Register will exist (other than adult Phase 1 studies). In the UK, any favourable opinion given by a research ethics committee is subject to the condition that the clinical trial is registered on a publicly accessible database. The time frame for publishing the summary of results is within 6 months of the end of trial for paediatric clinical trials or within one year of the end of trial for non-paediatric clinical trials. You do not need to submit this clinical trial summary report to the MHRA as well; however, you must send a short confirmatory email to CT.Submission@mhra.gov.uk once the result-related information has been uploaded to the public register and provide a link.
Pharmacovigilance
Guidance on qualified person responsible for pharmacovigilance (QPPV) including pharmacovigilance system master files (PSMF) from 1 January 2021
From 1 January 2021, the following legal obligations will apply to holders of UK marketing authorisations (MA). These include those that cover the whole of the UK, or are specific to Northern Ireland or to Great Britain (England, Wales and Scotland):
To operate a pharmacovigilance system for UK authorised products.
To have an appropriately qualified person responsible for pharmacovigilance (QPPV) that resides and operates in the EU or the UK and is responsible for the establishment and maintenance of the pharmacovigilance system for UK authorised products.
To maintain and make available upon request a pharmacovigilance system master file (PSMF) that describes the pharmacovigilance system for UK authorised products. The PSMF must be accessible electronically or physically from the UK at the same site at which reports of suspected adverse reaction may be accessed.
Statutory guidance concerning the QPPV for UK authorised products is described in the Good Pharmacovigilance Practices (GVP) Module I. This guidance will be supplemented by the ‘Exceptions and modifications to the EU guidance on good pharmacovigilance practices that apply to UK marketing authorisation holders’, which will be published in due course.
New guidelines have been outlined for Marketing Authorisations, to include Conditional MAs, registering new packaging information, guidance on the handling of applications for Centrally Authorised Products (CAPs), Article 29 applications, converting parallel distribution notices to UK parallel import licences, handling of ASMFs and CoS from January 2021, reference medicinal products, converting CAPs to UK MAs, guidance on licencing biosimilars, bioequivalence/therapeutic equivalence studies and renewing marketing authorisations.
New Submission Registrations
For planned applications for submission to the UK (for example, a Marketing Authorisation for the UK market), you will need to submit the information through the MHRA national portals.
All current Eudravigilance Gateway users who wish to gain access to the new MHRA Gateway will need to first gain access to MHRA Submissions. The steps for gaining MHRA Gateway access are contained within MHRA Submissions. MHRA Submissions will not be used to send or receive ICSRs.
If you need any clarification or support to help you to navigate the end of transition period please contact us and Ivowen will gladly assist you in a timely manner.
Written by Mary Canning
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-11-06 12:15:002023-11-06 11:15:33BREXIT – MHRA post-transition period information
Ivowen have provided some practical guidance and documents below, which may help you prepare for Brexit for products authorised by the DCP and MRP procedures.
Do you still need to change your MAH(s) in any of the EU Member States because of Brexit?
If so, refer to the following updated guidance issued in January 2019 which details the documentation and requirements to submit a Marketing Authorisation Holder transfer:
If you need any clarification or support to complete a Marketing Authorisation Holder transfer, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Is the UK your current RMS?
If so, you need to initiate an RMS switch as soon as possible. The guidance issued in July 2018 along with the template issued in June 2018 is required to complete this process. This template needs to be completed and sent to a specific e-mail address at the proposed new RMS. The list of contacts points to send the completed template is provided below:
If you need any clarification or support a RMS switch, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Do you need to change your UK batch release site, etc., if it currently within the UK?
If so, you can refer to the updated guidance document issued in December 2018, which provides information on the type of variation that is required to change functions such as batch release site(s), QC testing site(s), packaging site(s), deletion of site(s) for batch release and changes to the QP for Pharmacovigilance (QPPV) and PSMF where these functions still reside within the UK:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Do you supply product to Cyprus?
If so, the Cypriot Competent Authority has issued a newsletter to Stakeholders on the 14 January 2019 which lists 105 products authorised via an exceptional marketing authorisation affected by Brexit which are considered critical. The Drugs Council in Cyprus recommends that pharmaceutical companies register these products via an exceptional marketing authorisation using another reference state other that the UK.
If you need any clarification or support to complete an Exceptional Marketing Authorisation to avoid no supply of these critical products on the Cypriot market, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
The Health Products Regulatory Authority (HPRA) of Ireland is geared up and ready to support MA holders as they prepare for Brexit.
What happens if UK leaves the EU with no deal?
The implications of Brexit with regard to the UK’s role in the licensing of medicines will be determined by the terms of the ongoing exit negotiations. However, in accordance with Directives 2001/82/EC and 2001/83/EC:
For marketing authorisations issued through the mutual recognition procedure (MRP) or decentralised procedure (DCP), the Reference Member State (RMS) must be based in the EU/EEA.
The marketing authorisation holder (MAH) must be located within the EU/EEA.
MAHs will need to ensure that their EU qualified person responsible for pharmacovigilance (QPPV) and their pharmacovigilance system master file (PSMF) are located within the EU/EEA.
The batch release/testing site must be located within the EU/EEA.
HPRA are working “extraordinarily” hard on preparing for all scenarios
HPRA are Brexit ready
– Willing to act as RMS for all products where Ireland (IE) is currently CMS and a change of RMS is required. No fees will apply to the process for changing RMS to IE. The HPRA commits to an efficient and simple process for handling these requests (for example allowing the inclusion of multiple products in one request where applicable).
– To encourage the use of multilingual labelling the HPRA will proactively work with other European regulators to help optimise opportunities for multilingual labelling. The HPRA ‘Guide to labels and leaflets of Human Medicines’ has been updated (Oct 2018) to give specific guidance on the development of multilingual labelling
– HPRA MAH transfer procedures have recently been changed to allow MAHs up to 6 months to implement packaging changes following issue of the transferred authorisation, for Brexit related transfers. In addition the HPRA no longer requires stock to be recalled from wholesaler level six months following the issue of the transferred authorisation/ licence/registration.
Timeline for changing RMS
Can be completed within a matter of days. The critical issue will be the timing of when the change in RMS should occur as it is required to occur when there are no open regulatory activities for a product.
Procedures for MAHs to transfer MAH to EU/EEA based MAH
The transfer procedure must be used where the legal entity of an authorisation/licence holder is changed as marketing authorisations are transferred to a new company number. It is possible to transfer the MAH while there are ongoing/open variations. If the transfer is processed/issued, the new MA holder details will transfer onto the open/ongoing variations
Bulk transfers are accepted by the HPRA for Brexit and reduced fees apply.
New PA number is provided in advance of application.
Word version of an updated package leaflet is acceptable.
If the only changes proposed relate to MAH details and a new PA number, an Article 61(3) application is not required. However, if there are additional changes to the labels and package leaflet an Article 61(3) application must be submitted. This can be submitted in parallel to the transfer.
Medical devices containing an ancillary medicinal substance
Transfers of medical devices containing a medicinal ancillary substance with a valid CE certificate are considered administrative only. Transfers typically take less than 30 days from validation of the submission to be completed. No fees will apply to this transfer process.
Parallel Product Authorisations
If the UK is listed as one of a number of source countries on a PPA, this will need to be removed by way of a type IA variation (category 4).
Where the holder of PPA is located within the UK, the authorisation will need to be transferred to holder located within the EEA.
Qualified person certification of repackaging activity must take place within the EEA. Variations to change the site of batch certification can be submitted using the PPA variation category No 9a and 9b.
Variations to Marketing Authorisations to change Qualified Person Responsible for Pharmacovigilance (QPPV), manufacturing site or/and batch release sites.
Any variations required for a marketing authorisation (MA), e.g change in location of QPPV or site of batch release, should be completed prior to the date of the UK’s departure from the EU (prior to the 29 March 2019).
Changes in the QPPV, including contact details (telephone, and fax numbers, postal address and email address) may, for medicinal products for human use, be updated through the Article 57 database only (without the need for a variation)
Changes to the location of the PSMF (street, city, postcode, country) may be updated through the Article 57 database only (without the need for a variation)
Are you in search of advice and support on Brexit?
Please contact us, Ivowen are available to offer advice and support on HPRA compliance.
Written by Nanda Naik
https://eureg.ie/wp-content/uploads/2018/11/IE-EU-flag.jpg285390ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2018-11-21 16:10:012023-11-06 11:08:42HPRA’s support as you prepare for Brexit……
This year’s TOPRA Annual Symposium was held in Dublin in October. The Annual Symposium is an essential meeting for regulatory professionals to gain both an understanding of current and evolving regulatory requirements, as well as insights into future plans for regulations in the Human medicines, Veterinary medicines and Medical Device sectors.
Three members of our Team attended the Symposium this year, Majella Ryan, Alice D’Alton and Nanda Naik. This year was particularly satisfying for Ivowen as we had an exhibition stand at the three day event.
As well as exhibiting, Ivowen attended the sessions including ‘Life after Brexit’. This session was a live discussion of predictions and concerns about the possible outcome of ongoing negotiations with the EU.
Now that an extension has been granted, which will delay the UK exit until January 2020, we are left wondering if we will finally know the UK position when Ivowen attend the Medicines for Europe regulatory conference being held in Amsterdam on 29th – 31st January.
In the meantime, we await instruction from the regulators on whether UK will still be available on the CESP portal later this week. Ivowen is registered on the new MHRA submissions portal so either way we have you covered.
Ivowen will keep you up to date in the coming days and weeks
Please contact us, Ivowen are dedicated to keeping you up to date with the latest regulatory updates and innovations. We remain at your disposal to assist in all of your regulatory endeavours today and into the future.