
Back to Basics – Biological Medicines
Let’s get Back to Basics – Biological Medicines
According to Part I of Annex I of Directive 2001/83/EC, a biological medicinal product is a product that contains a biological substance.
A biological substance is a substance that is produced by or extracted from a biological source and that needs for its characterisation and the determination of its quality a combination of physico-chemical-biological testing, together with information about the production process and its control.
For example: recombinant proteins, monoclonal antibodies, medicinal products derived from human blood and human plasma, immunological medicinal products and advanced therapy medicinal products (ATMP) should be considered biological medicinal products.
Such biological sources can be bacteria, yeast, plant/animal cells, blood products, plasma-derived products, allergens and products manufactured using recombinant technology.
Biological medicinal products were previously developed mainly for rare diseases, but these days more of them are directed towards treatment of common diseases like diabetes, arthritis, inflammatory bowel diseases, asthma and cancer.
Unlike traditional small-molecule drugs manufactured by chemical synthesis, biological medicinal products are typically administered via injection or infusion requiring strict aseptic conditions and are manufactured through a highly complex, intricate & sensitive biotechnology process known as biomanufacturing.
RA professionals must navigate complex regulatory frameworks when submitting dossiers to seek product approval and to manage post-market compliance for these biological medicinal products.
Many of these products as well as ATMP medicinal products will require approval via the centralised procedure. Others (e.g. naturally derived biologicals) may be nationally authorised in individual Member States.
European Regulatory Affairs Limited specialise in the navigation of regulatory frameworks, gap analysis, communication with regulatory authorities & the planning & submission of dossiers to obtain product approval and manage post-market maintenance via the Centralised, National & MRP/DCP procedures.
Biologicals encompass a huge & diverse area and this article strives to provide a basic overview of some of the basic elements, approval pathways, and highlight some check-lists from the EMA & MHRA to follow before submission to avoid some common validation errors during review.
ATMP (Advanced Therapy Medicinal Products): are products for human use, including gene therapy, somatic cell therapy and tissue engineered products. ATMPs may also incorporate, as an integral part of the product, one or more medical devices in which case they are referred to as “Combined ATMPs”.
ATMPs can be classified into three main types:
- Gene therapy medicines: these contain genes that lead to a therapeutic, prophylactic or diagnostic effect. They work by inserting ‘recombinant’ genes into the body, usually to treat a variety of diseases, including genetic disorders, cancer or long-term diseases. A recombinant gene is a stretch of DNA that is created in the laboratory, bringing together DNA from different sources
- Somatic-cell therapy medicines: these contain cells or tissues that have been manipulated to change their biological characteristics, or cells or tissues not intended to be used for the same essential functions in the body. They can be used to cure, diagnose or prevent diseases
- Tissue-engineered medicines: these contain cells or tissues that have been modified so they can be used to repair, regenerate or replace human tissue
Approval Pathways: ATMPs, biologicals and biosimilars can be submitted using the following pathways depending on the biotechnological process to be used, whether the active is new and for which indications & Member States the MAA applications are being sought
- National – Great Britain (England, Scotland & Wales): All ATMP, biologicals & biosimilar products must be submitted according to MHRA guidance through the MHRA portal. For applications for Northern Ireland the EU guidance must be followed.
- Mutual recognition procedure/Decentralised procedure: Biological medicines can be registered through the mutual recognition or decentralised procedures, provided that they do not fall within the Annex to Regulation (EC) No 726/2004; in which case the centralised procedure has to be followed. Some biosimilars may be approved at national level, such as some low-molecular weight heparins.
- Centralised Procedure: All ATMP, biologicals & many biosimilar products must be submitted through the Centralised Procedure. All medicines produced using biotechnology and those for specific indications (e.g. for cancer, neurodegeneration and auto-immune diseases) must be approved in the EU via the centralised procedure. Nearly all biosimilars approved for use in the EU have been approved centrally, as they use biotechnology for their production.
Early Engagement with the relevant Regulatory Authorities is strongly recommended. The guidance should be thoroughly reviewed during development of a biological medicinal product & well in advance before the preparation of a MAA to understand what requirements are to be fulfilled.
Biosimilars: A biosimilar is a biological medicine highly similar to another biological medicine already approved in the EU (called “reference medicinal product”) in terms of structure, biological activity and efficacy, safety and immunogenicity profile (the intrinsic ability of proteins and other biological medicines to cause an immune response).
Developers of biosimilars are required to demonstrate through comprehensive comparability studies with the ”reference medicinal product” that:
- their biological medicine is highly similar to the reference medicine, notwithstanding natural variability inherent to all biological medicines
- there are no clinically meaningful differences between the biosimilar and the reference medicine in terms of safety, quality and efficacy
Biosimilar development relies heavily on comparability studies to establish similarity to the reference product. This involves a comprehensive head-to-head comparison of the biosimilar and the reference medicine.
Guidelines on Biologicals are outlined on the EMA & MHRA websites to facilitate the preparation of these Marketing Authorisation Applications (MAA). The guidelines on the EMA website for the active substance and the finished product are important sources of information when preparing MAA applications for these products.
The ICH guidance M4 (R4) on common technical document (CTD) for the registration of pharmaceuticals for human use, needs to be followed for the preparation of a well-structured Common Technical Document for applications that will be submitted to regulatory authorities.
Bio-processing:
This typically involves 2 stages
- Upstream
This is the stage where cells or microorganisms are prepared, cultivated and controlled so they can produce the desired biological output. It includes the preparation of a Master Cell Bank & from it a Working Cell Bank.
- Downstream
Downstream processing aims to isolate, purify, and concentrate the previously synthesized drug substance (DS).
Other critical steps in bio-processing are as follows:
Formulation & Fill-Finish: Final formulation to ensure stability, followed by sterile filling into vials or syringes in aseptic conditions to which regulatory authorities focus strongly on.
Quality Control (QC) & GMP Compliance:
-
- Process Validation: Documenting that the process consistently produces the intended result.
- Strict Documentation: Compliance with GMP, including detailed, auditable records for batch release.
- Environmental Monitoring: Ensuring sterile environments & conditions
Typical Biotech Manufacturing process with relevant ICH guidance’s:
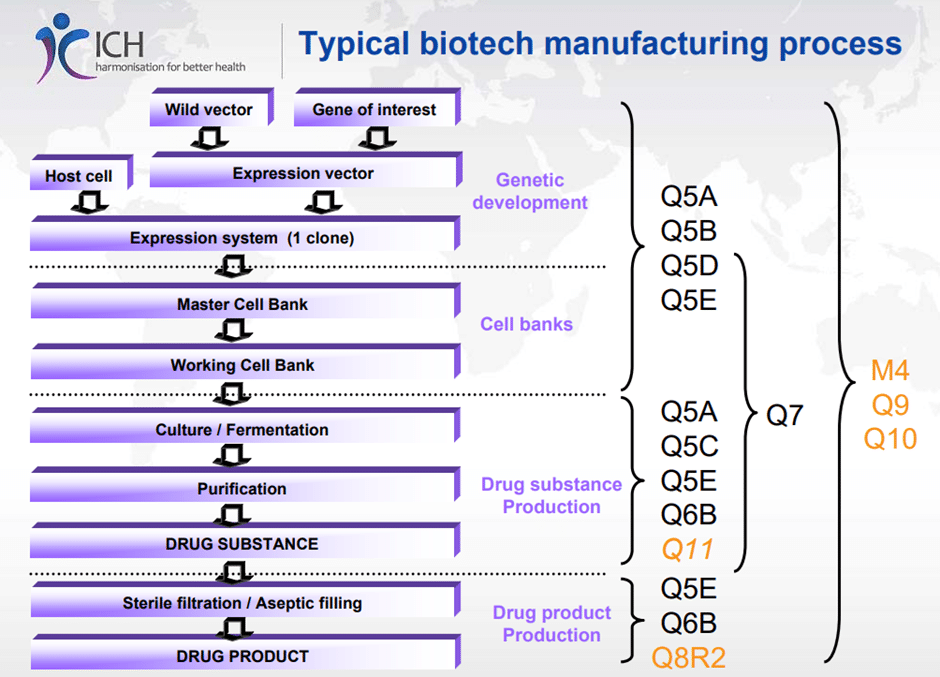
Figure 1 – source : ICH – Manufacturing Process for Biologicals
Check the EMA questions & answers guidance for biological medicinal products to ensure that all the required supporting documentation & reports are present so that CTD sections are complete, to pre-empt validation issues arising.
It is advised to review the EMA validation checklist before submission of the dossier and the MHRA Marketing Authorisation pre-submission checklist to keep validation issues to a minimum to help speed up review time.
If you would like to discuss any of the above and if you need support for any of your Biosimilar applications, we are ready to assist you and your team.
Feel free to contact us and we will be happy to help.
Written by European Regulatory Affairs Limited
Biotechnology: Technology that relies on biological systems, living organisms or components from living organisms (such as genes or enzymes) to make a specific product. A medicine obtained by biotechnology often has been produced by inserting a gene into cells so that they can produce the desired protein.
Comparability: Head-to-head comparison of a biosimilar with its reference medicine to rule out any significant differences between them in terms of structure and function. This scientific principle is routinely used when a change is introduced to the manufacturing process of medicines made by biotechnology, to ensure that the change does not alter safety and efficacy.
Immunogenicity: as defined by the European Medicines Agency (EMA), refers to a biological medicine’s ability to trigger an unwanted immune response, leading to the development of anti-drug antibodies (ADAs). These antibodies can cause adverse reactions, reduce drug effectiveness, or neutralize the drug’s therapeutic function.
MCB (Master Cell Bank): An aliquot of a single pool of cells which generally has been prepared from the selected cell clone under defined conditions, dispensed into multiple containers and stored under defined conditions.
Monoclonal Antibodies: Monoclonal antibodies are immunoglobulins (Ig) with a defined specificity derived from a monoclonal cell line. Monoclonal antibodies may be generated by recombinant DNA (rDNA) technology, hybridoma technology, B lymphocyte immortalisation or other technologies (e.g. display technology, genetically engineered animals).
WCB (Working Cell Bank): The Working Cell Bank is prepared from aliquots of a homogeneous suspension of cells obtained from culturing the MCB under defined culture conditions.
