![Feature Image [CoPilot]](https://eureg.ie/wp-content/uploads/2026/04/Feature-Image-CoPilot-1030x687.png)
Post Approval Change Management Protocols (PACMP)
So what is a Post Approval Change Management Protocol (PACMP)
The post approval change management protocol (PACMP) was introduced in the EU through the European Commission Guidelines on variation categories.
It is a regulatory tool which allows the management of specific quality change(s) in a more predictable and efficient manner, during the lifecycle of a product. Further details on PACMP are included in the ICH Q12 guideline on pharmaceutical product lifecycle management.
This regulatory tool is very useful for the implementation of multiple quality changes that an applicant plans to apply during the lifecycle of the product e.g. addition of an alternative manufacturing site, changes in a manufacturing process, changes in analytical procedures & changes in specifications.
It should be noted that a PACMP can only be submitted for a quality change which does not require clinical and/or non-clinical data assessment and does not result in a line extension.
There are two steps involved:
- The first step involves outlining the description, justification and a strategy to implement the changes in a protocol format (PACMP) which is submitted and assessed by the appropriate regulatory authority.
- The second step involves the submission of the actual variation(s), which contain the supporting data to implement the changes proposed and agreed in the approved PACMP.
Usually, a variation category designated for reporting changes under a PACMP (step 2) is at least one category lower than it would normally be (e.g. Type IB instead of Type II).
The aim of this stepwise approach is that faster and more predictable implementation of post-approval changes are obtained.
Advantages of submitting a PACMP
- Faster approval time – can reduce lead time to implement a change in the supply chain
- Provide predictability – no sudden surprises (all changes are agreed up front in the PACMP between the MAH and Regulatory Authority)
- More efficient than the alternative method of submitting numerous and various categories of variations which will not have been discussed or agreed upon from the start of the initiation of the change(s) – reduces risk of rejected variations
Submission & content of a PACMP
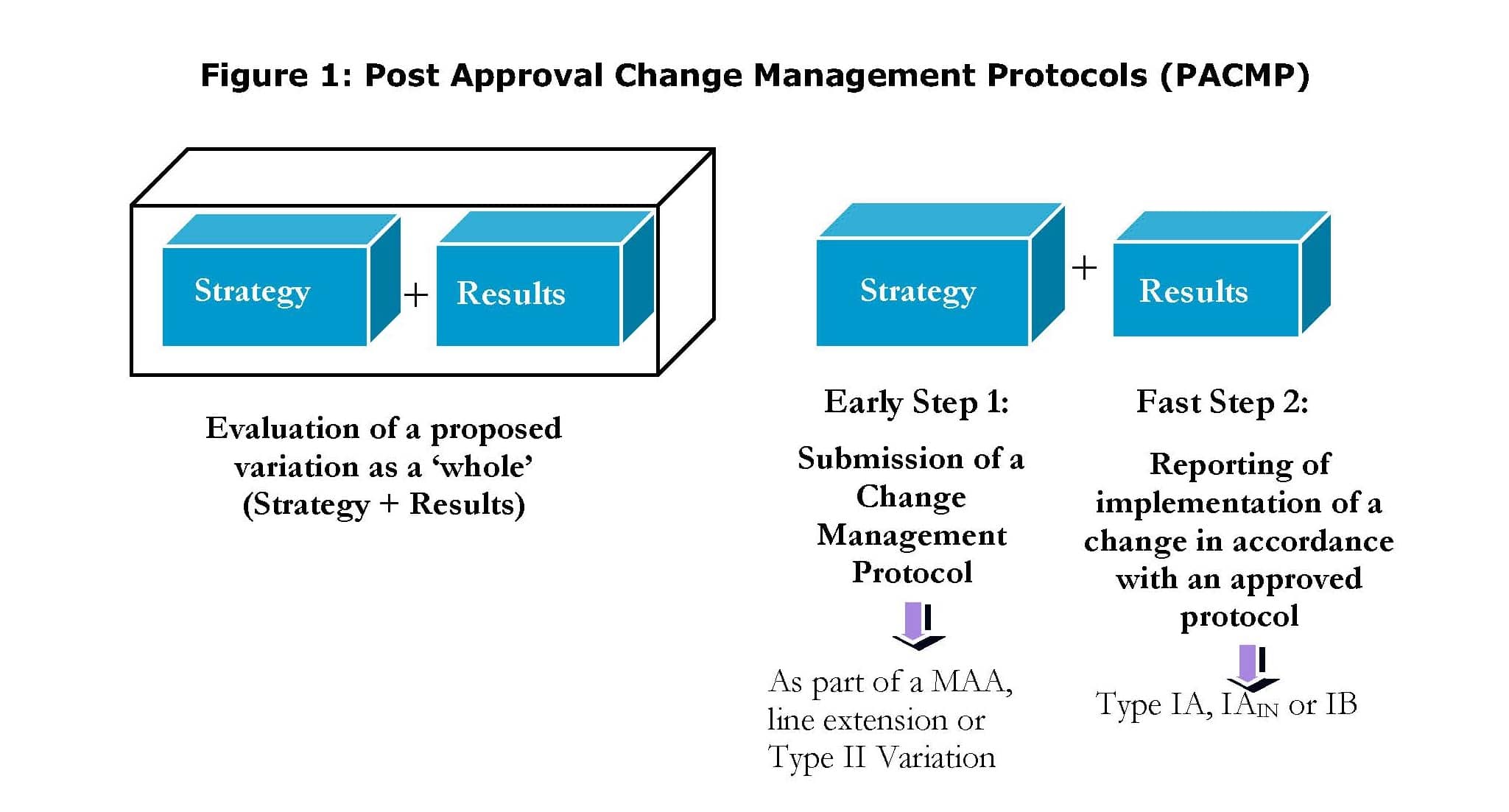
The application of a PACMP process typically involves the following two steps:
[source – EMA – Questions and answers on post approval change management protocols (PACMP)]
Step 1: Submission of a PACMP
- A PACMP may be included in an initial marketing authorisation application (MAA), a line extension application or may be submitted subsequently as a stand-alone Type II variation.
- The Variations Classification Guideline includes specific scope(s) for introduction of (Q.I.e.2, Q.II.g.2), change to (Q.I.e.4, Q.II.e.2) and deletion of (Q.I.e.3, Q.II.g.3) a PACMP for a drug substance and drug product respectively
- Organisations should review any planned PACMP(s) to confirm that they still align with the revised EMA guidance and the updated EU Variations Regulation
- The content of the protocol should include some of the following information as outlined in the guidance, depending on the nature of the change(s):
- A detailed description of the proposed change(s), including a rationale. The differences before and after the proposed change(s) should be clearly highlighted
- Based on an initial risk assessment, a list of specific tests and studies to be performed to evaluate the potential impact of the proposed change(s), such as; characterisation, batch release, stability, in-process controls, etc.
- The PACMP should include an appropriate description of the analytical procedures and proposed acceptance criteria for each test or study
- Discussion regarding the suitability of the approved control strategy or any changes needed to the control strategy associated with the planned change(s)
- Any other conditions to be met, such as confirmation that certain process qualification steps will be completed before implementation
- Where applicable, supportive data from previous experience with the same or similar products related to development, manufacturing, characterisation, batch release, and stability to allow for risk mitigation
- Proposed reporting variation category for step 2 of the PACMP
- Confirmation, as appropriate, that ongoing verification will be performed under the company’s PQS (Pharmaceutical Quality System) to continue to evaluate and ensure that there is no adverse effect of the change(s) on product quality.
- In cases where monitoring of the impact on product quality, following implementation of the change(s), is required a summary of the quality risk management activities should be provided to support the proposed PACMP. If multiple changes are to be implemented these activities should address the potential risk from the cumulative effect of multiple changes and how they are linked.
Step 2: Reporting of implementation of a change in accordance with an approved protocol
- A variation, under category Q.I.e.5 or Q.II.g.5 “Implementation of changes foreseen in a post-approval change management protocol (PACMP)” should be submitted to support the variations laid out in the approved PACMP, along with all required supporting documentation
Location of PACMP in the Dossier
A PACMP document should be located in CTD Module 3.2.R/2.3.R. When submitting the implementing variation, the relevant Module 2 and Module 3 sections, have to be updated.
Changes to a PACMP
If changes are required to be made to a PACMP after it is approved, the change can be submitted according to the appropriate variation category, as outlined in the current Variations Guideline.
Suggestions to improve finalisation of a PACMP
It is recommended that the applicant gets the supporting documentation finalised in a timely manner, so that it is ready for the submission of the implementation of the supporting documentation at step 2 of this process.
European Regulatory Affairs Limited specialise in the navigation of regulatory frameworks, gap analysis, communication with regulatory authorities & the planning & submission of regulatory approvals via the Centralised, National & MRP/DCP procedures.
If you would like to discuss any of the above and if you need support with your applications, we are ready to assist you and your team.
Feel free to contact us at info@eureg.ie and we will be happy to help.
Written by
Marian Winder

Marian Winder 1