You need to apply Type IA Annual Reporting in the EU in 2025
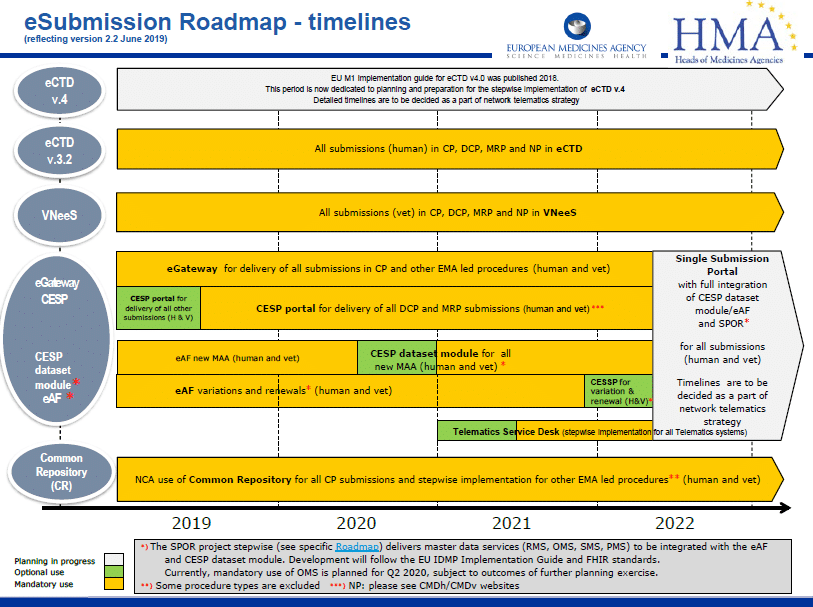
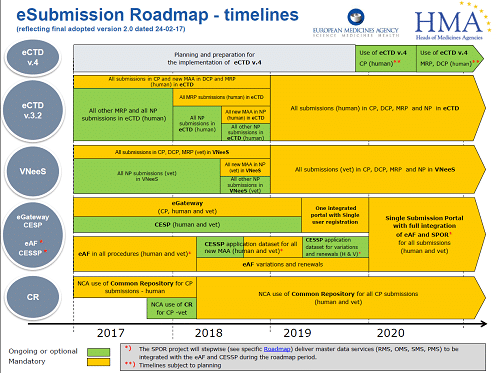
The Type IA Annual Update, also known as the “Annual report”, is a single submission of all the Type IA variations (not requiring immediate notification) which have been implemented during the previous twelve months.
The Annual report should be submitted as close to 12-month deadline as possible, but ultimately no earlier than 9 months and no later than 12 months after the first implementation date of the Type IAs included in the report.
Meaning of implementation for type-IA variations:
For quality changes, ‘implementation’ is when the company makes the change in its own quality system.
This implementation date must be carefully planned and recorded to facilitate submission of your Annual Reports.
How to plan the submission of your Annual Report:
As submission of the Annual report is dependent on the implementation of the first type IA variation, its submission date can change every year.
It is very important to note that the Type IA Annual report must fulfil the variation conditions for grouping (or super-grouping if it concerns more than one marketing authorisation).
It is expected that Type IA variations included in an Annual report will not be rejected. However, if rejected, Type IA variations from the annual report can be resubmitted as individual Type IAs immediately (outside of an annual report).
CMDh confirmed that although not specified in their guidance, Type IA variations implemented before 1 January 2025 can still be submitted as single type IA variations according to the current rules. The EMA confirmed that Type IA variations implemented in 2024 and not submitted to the Agency by 31 December may also be submitted by MAH no later than 12 months after implementation.
Therefore, Type IA variations implemented from 1 January 2025 should be submitted as part of the Annual report.
Type IAs can still be submitted outside the Annual report in the following cases:
- If they are part of a grouped Type IB or Type II variation
- If they are part of a super-grouping variation
- If it involves the resubmission of a Type IA variation previously refused in the Annual report (12-month reporting period is criteria in this situation)
- In exceptionally cases which should discussed and agreed with the EMA/NCAs.
eAF:
There are no current plans to amend the eAF to reflect Annual Reporting, instead, Applicants are advised to include a note in the cover letter and an additional note in the eAF scope to clarify that the application relates to the Annual Update of Type IA variations e.g. as foreseen in the updated Cover letter template:
<Annual update of type IA variation(s)
[X] We confirm that the annual update is submitted within 12 months following the implementation of the first type IA variation applied for in this notification. Implementation date of the first type IA variation: >
If you need any assistance with planning and submission of your Annual Reports, contact us and we will be happy to help.
Written by
Fiona Downey

Fiona Downey 1