This means that the MHRA do also require the submission of Annual Updates for Type IA variations, and these should be submitted nationally, unless the Type IA variations are submitted as part of a group with other variations applied for via the International Recognition Procedure (IRP).
IRP Variations:
You can use IRP during the lifecycle of UK products that have been initially authorised or subsequently varied via standalone;
National route
Decentralised and mutual recognition reliance procedure (MRDCRP) route
European Commission (EC) Decision Reliance Procedure (ECDRP) route
Conversely, where a product has been authorised via IRP, it is acceptable to submit standalone national post-authorisation procedures, including variations.
Variations submitted via IRP should be classified according to MHRA guidance on variations to MAs. To facilitate lifecycle management of the MA, variations should be submitted as soon as possible after approval by the Reference Regulator (RR).
The MHRA will be publishing updated guidance on Annual Updates very shorty. So, watch this space for a more detailed overview when it becomes available.
Written by
Fiona Downey
Fiona Downey 1
https://eureg.ie/wp-content/uploads/2023/10/Training-2.jpg306628ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2025-05-13 09:40:452025-05-13 09:40:45Type IA Annual Reporting in the UK
The Windsor Framework changed the regulation of medicinal products that were formerly in the scope of the EU centralised procedure so that these products are now licensed UK-wide under UK law.
Products not within this scope are authorised UK-wide under UK and applicable EU law (as amended by Regulation (EU) 2023/1182 and Directive (EU) 2022/642), as has been the case since 1 January 2021.
As a result, UK authorised products are now placed into one of two Categories:
Category 1 are UK products which fall under the mandatory or optional scope of the EU Centrally Authorised Procedure.
Category 2 are UK products which do not fall within the scope of Category 1 will be Category 2 products.
To assist MAHs in verifying the category of each of their products, a decision ladder has been created by the MHRA. The MHRA has also publish lists of Category 1 products and Category 2 products.
Which category your product falls under, will decide the pharmacovigilance requirements of that product.
Category 1 will be subject to UK legislation only. These products will legally be required to follow Part 11 of the HMRs for pharmacovigilance.
Category 2 will be subject to UK and EU requirements as applicable. These products will be legally required to follow Part 11 of the HMRs for pharmacovigilance with further pharmacovigilance requirements outlined in Schedule 12A of the HMRs.
However, it should be note that all existing exemptions that apply to all UK medicines remain in place irrespective of Category.
The Risk Management Plan (RMP) documents the risk management system considered necessary to identify, characterise and minimise a medicinal product’s important risks.
The RMP should be updated throughout the life cycle of the product, for example;
if there is a change in the list of the safety concerns
when there is a significant change in the existing additional pharmacovigilance activities
if an emerging safety issue is confirmed.
The RMP should be assigned a new RMP version number and a date each time it is updated and submitted for assessment.
An updated RMP can be submitted as a standalone [currently C.I.11] variation, when necessary, but it is most often submitted as a consequence of or part of another variation.
The format and content of the RMP should follow the RMP template; Guidance on the format of the risk management plan (RMP) in the EU – in integrated format which consists of seven parts.
For a generic medicinal product, hybrid products and/or fixed combination products with no new active substance the safety concerns should be aligned to those of the originator product that are available;
either from the originator’s approved RMP
or from the list of safety concerns of the substance published on the CMDh website.
RMPs are approved per each medicinal product, not per MAH, therefore only one RMP should be submitted for an MRP or DCP with different MAHs. The RMP must remain identical in RMS/all CMS throughout the product life cycle.
The RMP should be provided in eCTD section 1.8.2. To facilitate the assessment a ‘tracked changes’ version of the RMP, in Microsoft word, should also be provided in the ‘working-documents’ folder outside eCTD.
The QPPV’s actual signature or the evidence that the RMP was reviewed and approved by the QPPV should be included in the finalised approved version of the document; for eCTD submissions this would be the RMP submitted with the last eCTD sequence of the procedure (usually the closing sequence).
Feel free to contact us here at ERA to assist you with all things regulatory in Ireland, UK and across the EU.
We take the pain out of regulatory so that you can take your medicine to the next level.
Written by
Fiona Downey
Fiona Downey 1
https://eureg.ie/wp-content/uploads/2014/08/pharma-vigilance.jpg262228ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2024-07-19 09:53:292024-07-19 09:53:29Back to Basics – The Risk Management Plan (RMP)
The HPRA is now moving towards a system of slot allocation (expected submission dates) for newnational product registration applications, in addition to the existing DCP slot requirement.
Upon request, applicants will be given a submission slot in an agreed month.
This will help capacity planning for timely assessment of applications and will help maintain access for products on the Irish and European markets.
Slot-booking for national product authorisation applications
Applicants should contact RMS@hpra.ieno later than two months before their preferred submission date to request their preferred slot. The email subject title should include ‘National MA submission’.
The following documents should be provided when requesting a slot.
A CMDh common request form for reference Member States (RMS) which is available on the CMDh website.
A justification of the relevance of the application to the Irish market.
This should make reference to the unmet medical need your product will address and you may be asked if you intend to market the product in Ireland as part of your justification for applying to HPRA.
It is important that dossiers are complete before making a request, as delayed submissions may result in the loss of a slot.
If your preferred slot is not available, the next available slot will be allocated within a six-month window. Applicants will receive an email confirmation of a successful slot booking. While the HPRA are adapting to this new process we will try to facilitate applications which require an earlier start date.
Slot-booking for DCP authorisations
If you would like the HPRA to function as an RMS, please contact RMS@hpra.ieat least three months before your preferred submission date to request your preferred slot. The email subject title should include ‘DCP submission’.
When requesting a DCP slot applicants should also provide the completed CMDh common request form for an RMS.
Again, It is important that dossiers are complete before making a request, as delayed submissions may result in the loss of a slot.
If your preferred slot is not available, the next available slot will be allocated, within a two-year window. Applicants will receive an email confirmation of a successful slot booking. The HPRA also operates a cancellation list for slots which become available at short notice.
Scientific advice
Due to the complexities of Article 10a applications, applicants should obtain national scientific advice before submitting requests for such new applications. Further information can be found on our national scientific and regulatory advice page.
Ireland as a CMS
Furthermore, IE routinely participates as a Concerned Member State (CMS) in standard DCP procedures. A zero-day procedure as a CMS may be possible for medicines where Ireland is experiencing a critical shortage of a particular medicine.
Need support
Feel free to contact us here at ERA to assist you with all things regulatory in Ireland, UK and across the EU.
We take the pain out of regulatory so that you can take your medicine to the next level.
Written by
Alice D’Alton
Alice Dalton 1
https://eureg.ie/wp-content/uploads/2018/11/IE-EU-flag.jpg285390ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2024-03-28 12:06:472024-03-28 12:06:47Applying for Marketing Authorisation via national route or DCP procedure in Ireland – HPRA slot booking
Batches of medicinal product, authorised through the centralised procedure (CAP), that are currently lawfully on the market in Northern Ireland may remain so until their Expiry Date
However, these medicinal products cannot be moved from Northern Ireland to an EU/EEA Member State or be placed on the market in an EU/EEA Member State.
No new batches of authorised CAPs can be released on the market in Northern Ireland after Regulation (EU) 2023/1182 becomes applicable (01Jan2025)
Therefore the release of CAP medicinal product by your Qualified Person (QP) has to be planned accordingly and cannot be performed after this date.
It is recommended that companies and MAH’s anticipate any potential issues and resolve them in the next 8 months.
There is no possibility of multi-country packs for CAPs between Northern Ireland and any other EU/EEA member state after the regulation becomes applicable.
Any medicines placed on, or who are currently on the market before this happens may remain so until the expiry date is reached.
The product information (PI) of the CAPs will no longer require the mention of a local representative in Northern Ireland after the regulation becomes applicable.
The PI can be amended by way of an article 61(3) if no regulatory procedure is planned within 36 months.
This change should be made at the earliest opportunity.
The Q&As to stakeholders states ‘Additionally, it is also acceptable to request the deletion of the local representative for Northern Ireland in any regulatory procedure affecting the Annexes of the Marketing Authorisation submitted after 1 July 2024, provided that this change is only implemented after the date that Regulation (EU) 2023/1182 becomes applicable.’
Do you have a product that this may affect? There are lots of tasks that need to be done if so and these should begin this quarter!
The Legal Jargon behind it all
The Windsor Framework is a legal agreement between the European Union and the United Kingdom that was announced on February 27, 2023. It is a political declaration and a series of legal documents reached by the UK and EU in the Withdrawal Agreement Joint Committee. It covers topics such as trade, goods, services, sanitary and phytosanitary measures, and the democratic consent mechanism. The Framework was formally adopted by both parties on March 24, 2023, and came into effect on October 1, 2023.
Regulation (EU) 2023/1182 is a legal document that was passed by the European Parliament and the Council on June 14, 2023. It contains specific rules relating to medicinal products for human use intended to be placed on the market in Northern Ireland and amends Directive 2001/83/EC. Directive 2001/83/EC is aimed at ensuring that medicinal products placed on the market in Northern Ireland comply with the provisions of Union law until January 2025. The implementation of Regulation (EU) 2023/1182 has resulted in increased costs and red tape for GB-based pharmaceutical manufacturers and suppliers who are required to meet EU testing and labelling requirements in addition to UK rules.
Regulation (EU) 2023/1182 came into force on June 20, 2023 1. However, it is not applicable until January 1, 2025, provided that the UK has provided the written guarantees referred to in Article 8 of that Regulation.
This can impact centralised procedures (CAPs) as medicines can now only be placed on the market in Northern Ireland if authorised by the UK authorities in accordance with the law of the United Kingdom and under the terms of the authorisation granted by them.
Pursuant to Article 13 of Regulation (EU) 2023/1182, Article 5a of Directive 2001/83/EC is deleted with effect from the date on which the Regulation becomes applicable (01Jan2025). Article 5a allowed for the medicinal products to be temporarily authorised to supply to in Northern Ireland of a medicinal product belonging to the categories referred to in Article 3(1) and (2) of Regulation (EC) No 726/2004 provided that certain conditions were fulfilled.
If you need assistance with any of the above, contact us.
Written by Emily Fletcher
edited on 14/01/2025
Emily Fletcher 1
https://eureg.ie/wp-content/uploads/2024/02/Windsor-Framework-recd-13-02-24.jpg640960ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2024-03-19 15:57:312025-01-14 16:07:50UK Updates – Windsor Framework and Regulation (EU) 2023/1182 for Centralised Products
From the implementation of the Windsor Framework on 1 January 2025, the MHRA will regulate medicines across a UK-wide licensing regime.
Most new Marketing Authorisation Applications (MAAs) will be granted as PL licences covering the whole of the UK.
The MA number will have just a PL prefix (as it was before Brexit).
No action is necessary for this conversion for Marketing Authorisation Holders who only hold a PLGB licence for a particular product.
If a MAH currently holds both a PLNI and a PLGB for the same product and wishes to retain UK-wide authorisation, the PLNI will need to be cancelled in order for the existing PLGB to be converted to a PL.
Where an MAH only holds a PLNI and subsequently seeks authorisation for the whole of the UK, the PLNI will need to be cancelled prior to the granting of the PL.
MAHs involved in EU procedures, with NI included as a CMS, will need to notify the RMS to withdraw NI as a CMS from the procedure. This must occur before the cancellation of the PLNI and the granting of the new PL.
‘UK Only’ is required to be printed on the UK packaging from 01Jan2025, a sticker is allowed to be placed on packaging for a period of 6 months (until 30June2025) but this must be replaced with direct printing on the packaging after this.
And from 1 January 2025, the EU Falsified Medicines Directive (FMD) will no longer apply in Northern Ireland.
From 1 January features included for the purposes of compliance with EU FMD requirements may be removed or covered.
MHRA Overview:
Current position
From 1 January 2025
Action required from MAHs
New MAA for product in scope of the Centrally Authorised Procedure
GB: national applications, reliance/recognition applications,
PL
EC no longer able to authorise for NI.
For products entering the market from 1 January 2025, MAHs may apply for PL licences only. Must comply with labelling and packaging requirements.
New MAA for product in scope of the Centrally Authorised Procedure
NI: EC authorisation.
Supply of GB authorised products may be permitted through routes, e.g., NIMAR, Reg 174, Reg 167.
PL
EC no longer able to authorise for NI.
For products entering the market from 1 January 2025, MAHs may apply for PL licences only. Must comply with labelling and packaging requirements.
New MAA for Non-CAP product authorisations
UK-wide authorisation or option to apply for PLGB or PLNI
PL
Or
PLNI through the MRP/DCP
For products entering the market from 1 January 2025, MAHs may apply for PL licences or PLNI only if using the MRP/DCP. Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
UK-wide licence
PL
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
Separate PLGB and PLNI for the same product.
Option A:
PL only
By 30 September 2024 the MAH should request that the MHRA cancel the PLNI, effective on 31 December 2024. PLGB licences will convert to PL by 1 January 2025.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
Separate PLGB and PLNI for the same product.
Option B:
Retain NI only through the MRP/DCP.
No action required from MAH.
PLGB will be cancelled by the MHRA on 31 December 2024 as cannot hold a PL and PLNI simultaneously for the same product.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
PLNI through the MRP/DCP, with no PLGB for the same product.
PLNI through the MRP/DCP only.
Where a MAH subsequently applies for a PL, the PLNI will need to be cancelled prior to the granting of the PL. The MAH should inform the RMS of its intention to withdraw NI as a CMS from the MRP/DCP.
Must comply with labelling and packaging requirements.
Products with existing authorisations granted before 1 January 2025.
PLGB only
PL only
No action required from MAH.
PLGBs will be converted to PL by 1 Jan 2025.
Must comply with labelling and packaging requirements.
CAP Bridging Mechanism
Permits supply of GB-licensed product in NI for up to 6 months if no equivalent available.
Not applicable as the MHRA will license novel/CAP medicines in NI through a UK-wide licensing route.
Not applicable.
Existing Stock in Existing Packaging on the market
Continues as currently.
Can continue to be supplied to patients until the product’s date of expiry in the territory for which the product was valid for supply prior to 1 Jan 2025.
Not applicable.
More on Windsor Framework in the weeks to come……
If you need any help to navigate this new min field of UK updates, don’t hesitate to contact us for support.
Written by
Emily Fletcher
Emily Fletcher 1
https://eureg.ie/wp-content/uploads/2024/02/Windsor-Framework-recd-13-02-24.jpg640960ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2024-02-13 13:24:342024-02-13 13:25:45UK updates to your Medicine – Windsor Framework and Regulation (EU) 2023/1182
There are now several routes to obtain a marketing authorisation in the United Kingdom (UK), Great Britain (England, Scotland and Wales) or Northern Ireland. The options available will be determined by the intended market and the type of application. In this article we will discuss the purely National procedure.
To help you to decide what type of license you will require, here is a brief explanation of the new types of MA you can obtain:
MA prefix for UK
Possible MA types
Territory
Leg & Guidance
PL
UK wide
Authorised for use in United Kingdom (Great Britain & Northern Ireland)
EU & MHRA rules apply
PLGB
GB
Authorised for use in Great Britain only (England, Scotland and Wales only)
MHRA rules apply
PLNI
NI
Authorised for use in Northern Ireland only
EU rules apply
One option you can pursue is the National procedure (a 150-day procedure) to obtain a marketing authorisation (MA) in the UK, Great Britain or/in Northern Ireland. The MHRA has introduced this accelerated procedure aimed at expediting the availability of medicines for patients in the UK and proposes to reach its opinion on marketing authorisation applications (MAAs) within 150 days of filing an application (excluding the time taken to provide further information or data required).
The accelerated assessment is available for all high-quality new MAAs for both new, and existing active substances, as well as orphan designations. Interested applicants should contact the MHRA in advance of submitting the application.
For medicines containing new active substances or biosimilar products, the MHRA encourages applicants to provide a summary of the dossier to share their intentions and to verify the new active substance status.
The pre-submission meeting offers the opportunity to discuss the arrangements for the UK Compliance Check (CC) on Paediatric Investigation Plans (PIPs). Additionally, it also offers the opportunity to enhance joint discussion with National Institute for Health and Care Excellence (NICE) Health Technology Assessment (HTA) evaluation process.
The MHRA will operate a ‘fixed submission date’ system to facilitate consultation with the Commission on Human Medicines (CHM) and will publish a set of dates to facilitate planning the submissions to coordinate with appropriate meeting dates of CHM. The submission slots will be linked to the dates of CHM meetings.
The assessment timetable will begin after the validation of the application. The assessment process will run in two phases totalling 150 days like so:
Phase I: completed 80 days after the clock starts. Issues that arose or requiring clarification from the initial assessment will be raised with the applicant and should be addressed within the clock off period of 60 days.
Phase II: commence on receipt of the applicant’s responses. The MHRA will provide a decision on the acceptability of the product by day 150.
If the MHRA refuses to grant the MA-based on advice from CHM, there is an opportunity for the applicant to request a review of the decision.
The conclusion of the assessment will lead to the publication of a UK Public Assessment Report for the product.
Here are some useful links to obtain further information:
If you need any clarification or support to help you to navigate the new post Brexit procedures, please contact us and Ivowen will gladly assist you in a timely manner.
https://eureg.ie/wp-content/uploads/2019/01/Brexit2.jpg413640ivowenadminhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgivowenadmin2021-04-21 13:25:462023-11-06 11:15:36New UK National Procedure – Expedited 150 day procedure
The UK has left the EU and the transition period after Brexit comes to an end this year.
The MHRA have issued new guidance for industry and organisations effective from 01st January 2021. From this date the MHRA will be the UK’s standalone medicines and medical devices regulator.
Areas covered in the new guidance include:
Clinical Trials
From 1 January 2021, for registering clinical trials, existing and established international registers will still be used, such as ISRCTN registry (UK), or ClinicalTrials.gov (USA), to ensure the public is aware of your trial. For trials involving both UK and EU sites a record in the EU Clinical Trials Register will exist (other than adult Phase 1 studies). In the UK, any favourable opinion given by a research ethics committee is subject to the condition that the clinical trial is registered on a publicly accessible database. The time frame for publishing the summary of results is within 6 months of the end of trial for paediatric clinical trials or within one year of the end of trial for non-paediatric clinical trials. You do not need to submit this clinical trial summary report to the MHRA as well; however, you must send a short confirmatory email to CT.Submission@mhra.gov.uk once the result-related information has been uploaded to the public register and provide a link.
Pharmacovigilance
Guidance on qualified person responsible for pharmacovigilance (QPPV) including pharmacovigilance system master files (PSMF) from 1 January 2021
From 1 January 2021, the following legal obligations will apply to holders of UK marketing authorisations (MA). These include those that cover the whole of the UK, or are specific to Northern Ireland or to Great Britain (England, Wales and Scotland):
To operate a pharmacovigilance system for UK authorised products.
To have an appropriately qualified person responsible for pharmacovigilance (QPPV) that resides and operates in the EU or the UK and is responsible for the establishment and maintenance of the pharmacovigilance system for UK authorised products.
To maintain and make available upon request a pharmacovigilance system master file (PSMF) that describes the pharmacovigilance system for UK authorised products. The PSMF must be accessible electronically or physically from the UK at the same site at which reports of suspected adverse reaction may be accessed.
Statutory guidance concerning the QPPV for UK authorised products is described in the Good Pharmacovigilance Practices (GVP) Module I. This guidance will be supplemented by the ‘Exceptions and modifications to the EU guidance on good pharmacovigilance practices that apply to UK marketing authorisation holders’, which will be published in due course.
New guidelines have been outlined for Marketing Authorisations, to include Conditional MAs, registering new packaging information, guidance on the handling of applications for Centrally Authorised Products (CAPs), Article 29 applications, converting parallel distribution notices to UK parallel import licences, handling of ASMFs and CoS from January 2021, reference medicinal products, converting CAPs to UK MAs, guidance on licencing biosimilars, bioequivalence/therapeutic equivalence studies and renewing marketing authorisations.
New Submission Registrations
For planned applications for submission to the UK (for example, a Marketing Authorisation for the UK market), you will need to submit the information through the MHRA national portals.
All current Eudravigilance Gateway users who wish to gain access to the new MHRA Gateway will need to first gain access to MHRA Submissions. The steps for gaining MHRA Gateway access are contained within MHRA Submissions. MHRA Submissions will not be used to send or receive ICSRs.
If you need any clarification or support to help you to navigate the end of transition period please contact us and Ivowen will gladly assist you in a timely manner.
Written by Mary Canning
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-11-06 12:15:002023-11-06 11:15:33BREXIT – MHRA post-transition period information
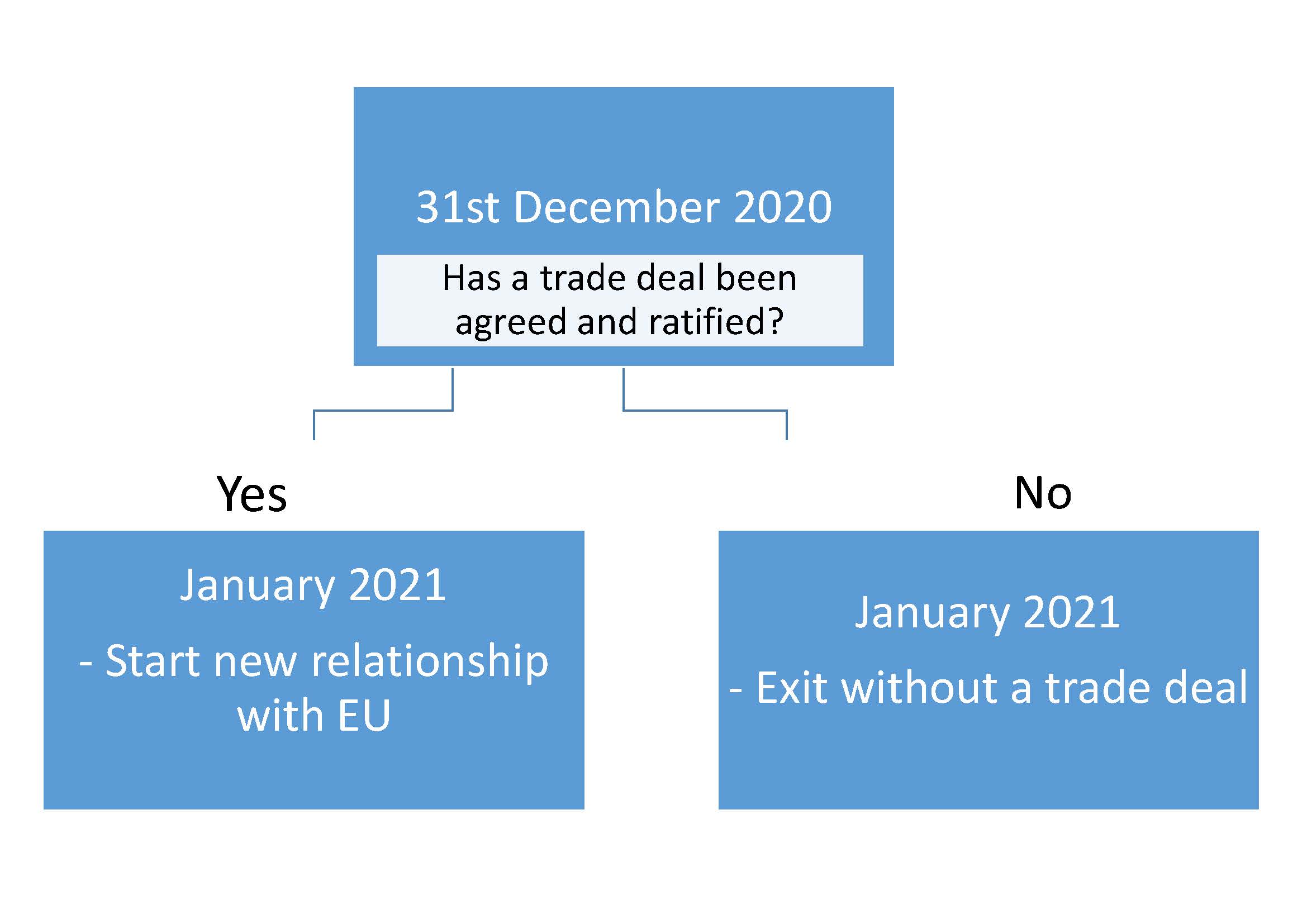
It seems that Brexit is on course for 1st January 2021….
The 30th June 2020 came and the legal deadline for agreeing to an extension of the transition period has passed with no request from the UK. Therefore the Brexit trade deal transition phase will come to an end on the 31st December 2020. At this stage there will be one of two possible outcomes:The next important dates are the 15-16th October (European Council Meeting) and 26th November (penultimate plenary session of 2020. European lawmakers have stated that a trade deal must be negotiated, checked, translated and presented to the European Parliament by this date if the transition period is to end by 31 December 2020). If the UK exits without a trade deal, trade between the UK and EU will change immediately on the 1st January 2021 (i.e. Hard Brexit).
The UK Government launched a major new public information campaign to give everyone the facts that they will need to be ready for 1 January 2021. A straightforward checker tool at gov.uk/transition will help identify some of the specific steps any business or individual needs to take to be ready, and will allow companies to sign up for bespoke updates.
If you require any assistance for UK products please contact us: info@eureg.ie
Written by Emily Fletcher
Emily Flecther
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-07-22 12:42:222023-11-06 11:08:37BREXIT – No extension to the transition period
In response to the significant impact the COVID-19 pandemic is having on European regulatory activity, the European Commission, the European Medicines Agency and the Heads of Medicines Agencies network (EC, EMA and HMA, respectively) have approved a number of measures to help the management of marketing authorisations for human medicinal products considered crucial during the pandemic period.
The objective of these measures agreed at European level is to promote regulatory flexibility, facilitate, simplify and accelerate the administrative procedures, as far as possible, in order to respond more efficiently to emerging needs during this period.
As a result, the EC recently published questions and answers on regulatory expectations for medicinal products for human use during the COVID-19 pandemic:
This Q & A document which provides guidance to marketing authorisation holders (MAH) includes the following topics:
renewal applications
sunset clause
an exceptional change management process (ECMP) for crucial medicines for use in COVID-19 patients
circumstances under which the validity GMP certificates and authorisations to manufacture/import can be extended
circumstances under which the validity GDP certificates and wholesale authorisations can be extended
adaptions to the work of a Qualified Person (QP)
the possibility of adapting quality requirements for medicines intended to be used for the treatment of COVID-19 patients
the impact on reporting into EudraVigilance of Individual Case Safety Reports (ICSRs)
flexibility in the labelling and packaging requirements to facilitate the movement of medicinal products within the EU
Further to the European Commission’s Q&A document, the CMDh has agreed additional questions and answers that provide practical information on how to specifically address and apply the provisions determined by the European Commission for MR/DC procedures:
The CMDh document addresses issues such as the impact of COVID-19 on assessment timelines, how to use the ECMP procedure (which is only applicable for products that are crucial for the treatment of COVID-19 patients) and QP declarations based on a desktop audits. It also includes a useful annex that details Member States’ email addresses and links to relevant published guidance on MS websites.
Both documents will be updated and supplemented with additional information, as appropriate during the pandemic.
Everyone at Ivowen is working tirelessly to keep our clients applications on track. We are liaising with the National Competent Authorities all the time to ensure we avoid delays and get the best results possible in these unprecedented times.
If you need any assistance in this regard please don’t hesitate to contact us.
Written by Claire Brown.
https://eureg.ie/wp-content/uploads/2020/05/Corona-virus-image-11-05-20-1.jpg405720ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-05-11 16:24:572023-11-06 11:15:46European Procedural Guidance during COVID-19 Pandemic